Michelanglo — Documentation
General implementation
This is a generalised instructions for implementing a view from Michelanglo on your site. For the code specific to your generated view see your pages and click the button "implement" on the side.
Raw HTML mode
First, you can only use the code if you have a website that you can edit as raw HTML. Otherwise, you can only share links or zipped html files.
Not necessarily of the whole page as only a small part is fine. For example:
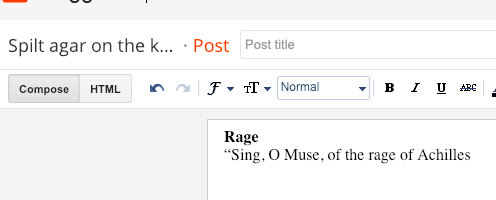
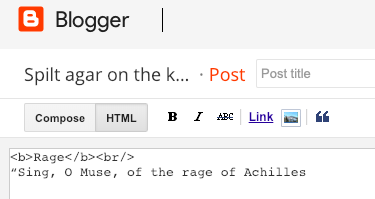
In the first case, the HTML code is hidden as one sees what one gets as an end result. In the second case, the HTML code is visible: words between tags such as <b> are not styled. In most cases JS can be added here.
If it does not work on your site, it may because some information is lost when you added it.
Try adding to your page:
I am definitely in the correct HTML editor mode as this is <b>enboldened</b> and this is <span id='blue'>blue</span>.
<script type="text/javascript">document.getElementById("blue").style.color = "blue";</script>
And view it.
- If the emboldened text is not bold, but has
>b<before it, you were ending your html page in an editor that showed you the end formatting (WYSIWYG) not the raw HTML code. - If the emboldened text was bold, but the ought-to-be blue text was not, they the editor may be stripping JS for security reasons or you switched from raw to WYSIWYG before saving and it stripped it.
- If both displayed as hoped then it is trickier.
On Chrome show the console. To do so press the menu button at the top right next to the your face, then "More tools..." then "Developer tools". Here you can see what went wrong with your page. Is there a "resource not found error"? If so, you may have set it to fetch something that was not there or in that location.
If the demo image gives you an unsolicited black, that means something went wrong with the parsing of the parts. See the else {return 0x000000} //black as the darkest error! line, which is there as a last ditch.
To debug this yourself, open the console and type protein.structure.eachAtom(function(atom) {console.log(atom.chainid);}); or in the inner bit atom.resno or other property of atom until you figure out what is wrong with your structure.
The variable protein can be obtained with stage.compList[0] if the stage is exposed or if you are using NGL extended script NGL.getStage('viewport').compList[0] or NGL.getStage('viewport').getComponentByType('structure').
If you thing, the fault is in the code please email me. I am aware of two unfixed bugs, one is the CD2 atom in histidine residues with different colored carbons and the other is the absence of shades of gray (_e.g._ `gray40`) in the color chart.
CDN
In order to make the JavaScript magic happen, the libraries need to be loaded.
Add to the bottom of the page, but before the closing of the body element, the following lines.
First, Michelanglo.js uses JQuery, so make sure it is present. Namely, if nothing works and in the console there is an error saying $ is undefined or if you add
<script>alert('JQuery absent') ? $ === undefined : alert('JQuery present')</script> into the document you get a negative.
<script src="https://code.jquery.com/jquery-3.4.1.min.js"
integrity="sha256-CSXorXvZcTkaix6Yvo6HppcZGetbYMGWSFlBw8HfCJo="
crossorigin="anonymous" type="text/javascript"></script>
Then you can add the michelanglo.js code:
<script src="https://unpkg.com/ngl@2.0.0-dev.34/dist/ngl.js" type="text/javascript"></script>
<script src="https://michelanglo.sgc.ox.ac.uk/michelanglo.js" type="text/javascript"></script>
Alternatively, host these two files in your server and link to them.
<script src="/ngl.js" type="text/javascript"></script>
<script src="/michelanglo.js" type="text/javascript"></script>
These scripts load NGL and allow it to be controlled via data-* attributes of HTML elements (see markup for more)
Also, if you are using Bootstrap 4 (buggy on 3) the button that shows options can be added with the following code placed after the other parts.
<script src="https://michelanglo.sgc.ox.ac.uk/michelanglo_menu.js" type="text/javascript"></script>
Viewport
Best option
The recomended way (simple/powerful) is have the viewport declared (in the correct location) like so<div role="NGL" proteins="[{...}]"></div>, where the JSON string value (=in quotes) of proteins is an array of objects like this: {type: 'rcsb|file|data', value: 'PDB id|path|multiline string', loadFx: myOptionalFancyLoadFunction}. Where the optional load function can be added to a script element and can be made from a PyMol file (see PyMol converter).
Full options
There are four inter-compatible ways to declare the viewport.
- old way. In JS you load it as the NGL suggests, but keeping the stage object globally declared as
stage:window.stage = new NGL.Stage( "viewport",{backgroundColor: "white"}); stage.loadFile('static/gfp.pdb')— addingstageas a property ofwindow(i.e.window.stage = …) is the same as declaring withoutvar(i.e.stage = …; cf.var stage = …). - Modded old way. As above but you add your stage to the object
NGL.stageIdthuslyvar id = "viewport"; NGL.stageIds[id] = new NGL.Stage( id,{backgroundColor: "white"}); NGL.stageIds[id].loadFile('static/gfp.pdb') - MultiLoader way. Using the
multiLoaderfunction described below (NGL.specialOps.multiLoader('viewport', [{type: 'rcsb', value: '1ubq', loadFx: myFancyLoadFunction}], 'gainsboro')), which allowsdata-loadprolinks to toggle between multiple proteins (pdb strings, custom pdb files or RCSB pdb entries) each with an optional load function. - No JS way. Using the attribute
role="NGL"on the viewer div-element in combination with prolinkdata-*attributes, e.g.<div id="viewport" role="NGL" data-load="1ubq" data-focus="residue" data-selection="30:A" data-view="[…]">. If multiple PDB proteins are required, usedata-proteinsas opposed to load where the value is a comma separated space free list of codes (not array), these can be accessed viadata-load=3prolinks as usual (this is less clear than simply specifying prolinks withdata-loadwith a file or PDB code, but has the sole benefit that, data-load='1ubq' will re-load the file even if 1ubq is loaded, while data-load=2 will not if the current index is 2, however in future unique names will be implemented to avoid this drama). Note that the height issue is sorted and that it can be combined like everything else with click-to-enable-NGL images.
Image
If you are adding an image (as described here), you might need to add it manually as many WYSIWYG editors with insert image buttons (e.g. Blogger) make images that when clicked result in a pop-up with the image fullsize, which is obviously incompatible.
Therefore add or edit the image thusly: <div id="viewport"> <img src="my_protein.jpg" alt="my protein" width='100%' style='cursor: pointer'></div>.
The CSS style can be different, but the important thing is that there is a width or a min-width and a height or a min-height —in
this example the 0 height is a special case and results in the height being equal to the width.
myData object and multiLoader
The load ability, which uses the add-on NGL.specialOps, works preferably in combination with the add-on NGL.specialOps.multiLoad,
which can initialise the scene and handles such things.
The data for load/multiLoader is stored in myData object, which has the property myData.proteins,
which is a list of {name: 'unique_name', type: 'rcsb' (default) | 'file' | 'data', value: xxx, 'ext': 'pdb' , loadFx: xxx}.
The optional argument loadFx is a function that accepts as argument a NGL protein (component) object and performs requested operations.
The MultiLoader and load can handle img elements in the div, namely the case where you start with an image with labels etc and you click on it and it switches to the viewer.
function nice_ubiquitin (protein) {
protein.addRepresentation( "line", {color: ..., sele: ...} );
bla bla
}
NGL.specialOps.multiLoader('viewport', [{type: 'rcsb', value: '1ubq', loadFx: nice_ubiquitin}], 'aquamarine')
//////////////////////////////id/////////array of elements with loadFx////////////// ////background
The data-load can load proteins from the myData.proteins (and run the custom function LoadFx) if an index is provided.